The Fukazawa et al. paper I
described before had a good deal of evidence suggesting that actin polymerizes in response to tetanic stimulation and underlies synaptic potentiation. They had the advantage interpretation-wise of working in live animals where the actin is remodelling in its natural environment. However, this comes with the disadvantage of not being able to observe the process directly. The hippocampus is buried deep in the rat brain and has to be dissected out and fixed in paraformaldehyde before it can be labeled with fluoresecent phalloidin (the polymerized actin indicator). The earliest timepoint after LTP induction we see in Fukazawa et al. is 20 minutes.
Okamoto et al. (
pdf) developed a novel approach using
fluorescence resonance energy transfer (FRET) to view actin's response to stimulation in cultured hippocampal tissue. This buys them the ability to be very certain about when and where (down to which compartment of the dendritic spine) these events occur. A drawback is that they have to use a model system that is now several steps away from the primary phenomenon of interest (memory): LTP instead of behavioral training and tissue that has developed in a dish lacking any external stimulation or feedback from other brain regions. Everybody does it, but before you see how pretty their FRET pictures are, you have to be reminded that they come at a price.
FRET is less complicated than it sounds. It's the fluorescence equivalent of billiard balls bumping or dominoes knocking each other over. In the past handful of years, molecular biologists have branched out and modified the green fluorescent protein (GFP) that we all know and love. Now there are
blue, yellow, red, and cyan fluorescent proteins and they each have characteristic excitation and emission wavelengths. The excitation wavelength is the specific color you can hit the molecule with to put it in an excited state. As the fluorophore relaxes back to its ground state it releases energy at its emission wavelength. In FRET a donor fluorescent protein emits energy that can excite an acceptor protein. So you shoot light in to excite the donor and,
if the donor and acceptor are close, you will get signal back out at the emission wavelength of the acceptor.
Okamoto et al. tagged actin monomers with CFP and YFP, excited CFP, and looked for YFP emissions as an indication that actin had polymerized. This is illustrated below:
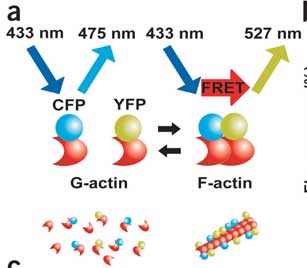
The authors first show that the FRET signal really is reflecting actin dynamics as an actin depolymerizing drug (latrunculin A) reduces FRET and an actin filament stabilizing drug (jasplakinolide) increases FRET. They go on to investigate the effects of various plasticity-inducing signals. The first pass was bath application of NMDA (the specific synthetic agonist that NMDA receptors are named after). They found a relatively rapid (within 3 minutes) increase in polymerization. This is a little troublesome because other papers that the authors cited have shown an decrease in F-actin (the polymerized form) following NMDA bath application. This decrease was attributed to excitotoxicity, where excess calcium influx can actually lead to cell death. This is thought to be a mechanism by which extensive brain damage occurs in the face of traumatic brain injury and oxygen deprivation. It could be that the previous studies and this one use different model systems or that the other studies didn't have the temporal resolution to observe this initial increase. Since you asked, I don't think people should bother bath applying neurotransmitter agonists in plasticity studies. Constant receptor occupancy is so far from the physiological norm it seems impossible to interpret this information correctly.
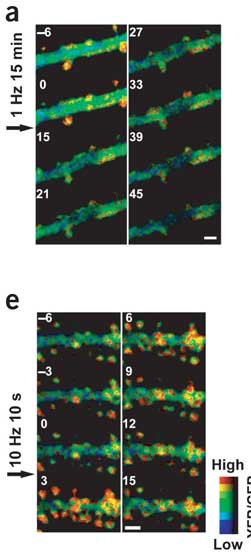
Fortunately, the authors went on and used common electrophysiological stimuli for the rest of the paper. It is still hard to say whether the activity induced by these protocols is like that in the brain, but at least neurons are deciding how much neurotransmitter is in the synapse instead of researchers dumping it on for 10 minutes at a time. One of the most remarkable findings is how fast the actin state changes in response to tetanic stimulation. In a particular spine-head, FRET levels increased within 20 seconds of stimulation. The increase is synapse-specific, meaning that spines that are on the same dendrite but distant from the stimulation electrode don't show a change. This shows that the signaling events necessary to induce actin polymerization must happen right there next to the spine rather than being a dendrite or cell-wide event.
I've written a lot about LTP, but its sinister partner never gets as much play. You can increase synaptic strength using high-frequency stimulation, but you can also decrease it with low-frequency stimulation. The lasting decrease is termed long-term depression (LTD). Okamoto et al. followed actin dynamics after a dose of high or low frequency stimulation. In accordance with the Fukazawa et al. findings, LTP increased actin polymerization. However, the increase was observed within 3 minutes. F-actin decreased after low-frequency stimulation, but it's harder to say how long it took because the stimulation took 15 minutes. There was a huge reduction in FRET signal from before the stimulation (0 min) to right after (15 min). This might be a good time to show you some FRET pictures. The big green thing is a dendritic branch. The little knobby-do's sticking off are dendritic spines. The top image is LTD stimulation, and the bottom is LTP. Actin polymerization is sort of orange. The numbers indicate minutes in relation to the start of stimulation.
What is the functional outcome of actin polymerization for a spine? For one, the spine head widens in correlation with the increased FRET signal. I've read enough to know that there is a pretty consistent relationship between spine head width and the number of AMPA receptors in a synapse, which makes sense. Another interesting consequence of actin polymerization that the authors noted was an increase in the beta subunit of calcium/calmodulin-dependent kinase II (CaMKII) in the post-synaptic density. I know that last bit is jargon-y. I say it "Cam kinase two beta". CaMKII has alpha and beta subunits that form together into a 12-unit holoenzyme that plays a structural and signaling role on the receiving side of the synapse, attaching to some receptors and phosphorylating others. I've gone into more detail about this particular molecule here.
The obvious step after this is to see if this plays out in an actual learning situation. Joe LeDoux has started in on this route, but I don't think he's measured polymerization directly. My friend Joe (not LeDoux) posted a little something on that a while back. Also, we know now that polymerization can lead to CaMKII movement, but is it a reciprocal relationship? Also, which comes first, CaMKII signaling, spine head widening, or AMPAR insertion? Or does it all happen in one big "whoosh"? Perhaps all of the events are initiated in parallel by an unspecified (probably calcium-activated) signaling event.
Addendum:
I forgot I wanted to mention that a second high-frequency stimulation could induce even more polymerization after the first burst. The authors contrast this with the electrophysiology which seems to potentiate in an all-or-none fashion. While it is true that LTP generally goes up to the same peak during induction, more stimulation can lead to longer-lasting LTP. So it strikes me that the amount of actin that accumulates in filaments in a spine head during initiation must determine duration rather than magnitude. If that's true then it must go for the other correlates of actin polymerization: spine head width, CaMKII accumulation, the presumed AMPAR insertion. But more AMPARs ought to create greater synaptic strength, so that ought to affect magnitude. I don't have the answer at the moment. One of these correlations needs to be disconnected.