The route of neurotransmission that you learn about in an introductory neuroscience class is action potential driven neurotransmitter release. A wave of depolarization travels down the axon to the bouton and activates voltage-sensitive calcium channels allowing calcium in to trigger vesicle exocytosis. Synapses get bored though, sitting around waiting for an action potential, so they dribble a little neurotransmitter across every now and then just for kicks, producing mini excitatory post-synaptic potentials (mEPSPs, minis). Actually, it is not clear what the purpose of mEPSPs is, but recent studies are indicating that they may serve some kind of homeostatic role, suppressing boundless synaptic growth and change. I should probably mention that all these studies that I know of relate to excitatory glutamatergic transmission, and of course AMPARs and NMDARs are the major receptor subtypes for this brand of neurotransmission (somebody tell me when repeating this factoid gets patronizing). The starting point for these investigations is the phenomenon of
synaptic scaling. When neurotransmission is pharmacologically blocked for some length of time the strength of synapses starts to crank up. One can observe this even just in the size of minis before and after blockade. What are minis doing? What processes are you disinhibiting when you remove them from the picture?
Back in 2004, Schuman and colleagues blocked NMDA or AMPA receptors or both and monitored synthesis of a GFP reporter flanked by the regulatory elements from calcium/calmodulin-dependent kinase II (
CaMKII). All of these mini-blocking manipulations led to marked increases in the reporter, with the NMDA receptor manipulation alone producing the weakest (~40%) increase. Other studies have indicated that minis regulate spine structure and post-synaptic signaling pathways like CaMKII and MAPK activation. This
2006 Schuman paper (Sutton et al.) is a technical tour de force providing evidence that minis are locally suppressing protein synthesis-dependent insertion of a special plasticity-related type of AMPARs that are later replaced with the more mundane (GluR2-containing) AMPARs. Many papers that have been concerned with spontaneous neurotransmission have blocked action potential firing, but this doesn't quite exactly get at the role of minis. Instead, Sutton et al. blocked action potentials along with glutamate receptors. Since AMPAR transmission provides the largest initial component of minis it is a little difficult to observe scaling of minis while an AMPAR blocker is around, so most of the experiments in this paper focus on the effect of NMDAR blockade + action potential blockade. The effect of this manipulation is to greatly accelerate synaptic scaling as compared to that observed for simple action potential blockade, suggesting that NMDARs serve to inhibit scaling.
Time for an AMPAR subunit review. Most AMPARs in the nervous system contain GluR2 subunits. Certain properties are associated with GluR2: a short cytoplasmic tail, calcium-impermeability, and resistance to blockade by polyamines. GluR1 and some other subtypes have the opposite characteristics, but you have to have a GluR2-lacking receptor before you can get calcium-permeability and polyamine-sensitivity. So GluR2s sit around in a synapse. Something happens to strengthen that synapse, and GluR1s are inserted. Then the GluR1s are slowly replaced by GluR2s again, but the synapse now has overall greater receptor capacity. Sutton et al. proceed step-by-step showing that NMDAR blockade (using a drug called APV) causes more GluR1 to be synthesized, produces more GluR1 on the cell surface, and produces more GluR1 specifically co-localizing with synaptic markers. They did not detect more surface or synaptic GluR2. There are more synapses containing GluR1 and more GluR1 signal at each synapse where it is found. It seems then that, in the absence of minis, GluR1 is being driven into synapses without GluR2.
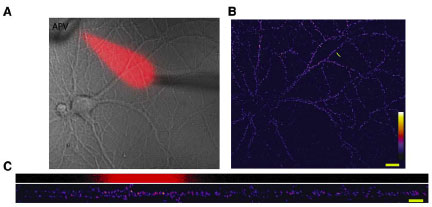
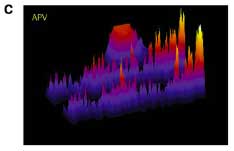
To get at the spatial specificity issue they had to be able to pharmacologically manipulate just a section of a dendrite, so they came up with this dual micropipette perfusion system. They put a marker in with the perfusate so you can see where the drug is going, producing a really nice visual:
So check it. The red ghost indicates the portion of the dendrite receiving APV. The signal scale in the pics on the right and bottom is for surface GluR1 expression. Effin' cool right? The effect of minis is local. They also inverted the logic and globally blocked NMDARs while locally blocking protein synthesis and got basically the opposite pic. This sez that some protein is being locally translated that is responsible for GluR1 insertion in the absence of minis. I suppose it could just be GluR1, but you sort of feel like there needs to be more scaffolding and whatnot.
To get at the identity of these new receptors a little more, they used a polyamine derivative that specifically blocks GluR2-lacking AMPARs. This reversed the accelerated scaling that occurs with NMDAR blockade, but not the scaling that occurs after 24 hours of simple action potential blockade. The mechanisms aren't entirely different though, by 24 hours the NMDAR-scaling is also mostly polyamine resistant. This result is consistent the slot hypothesis, and with the time course indicated by McCormack et al, 2006. GluR1s move into synapses to increase strength and bring a structural apparatus with them. Then the GluR1s are slowly exchanged with GluR2-containing AMPARs.
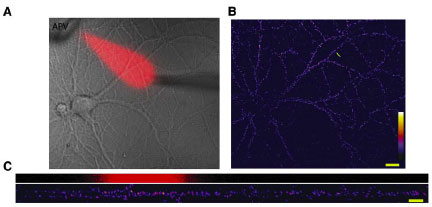
OK. I know you're getting tired, so just one more pretty picture and I'm out. Schuman and colleagues took advantage of one more property of GluR2-lacking AMPARs that I had no idea about. I really have no idea how they find this stuff. GluR2-lacking AMPARs are cobalt-permeable. There is a fluorescent molecule called calcein that is quenched (turned off) in response to cobalt. So we can track functional GluR2-lacking AMPARs by flooding the cell with calcein, and providing cobalt in the extracellular medium. They combined this with their local perfusion system to get to the final conclusion, the title of figure 7: NMDAR mini blockade induces local, protein-synthesis-dependent insertion of GluR2-lacking AMPARs. I'm gonna skip the protein synthesis part. It is about the same as above. Just look at these dendrites.

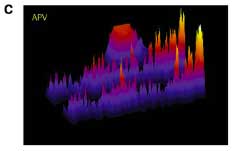
The top red blotch in figure B indicates the portion along the dendrite where APV is flowing. The top dendrite is before adding cobalt to the solution and the bottom is after. Figure C turns this 3D with signal intensity as the Z-axis. Note the specific decrease in signal after cobalt corresponding to the area of APV perfusion. Along with studies indicating that AMPAR transmission suppresses Arc transcription, I think an important new area is developing to focus on the steady-state processes that occur in neurons. The activity level of these processes may also play a role in determining the likelihood of future plasticity.
Related:
The Slot Hypothesis
BDNF and Arc regulation: NMDARs vs AMPARs