
Not quite, but interesting article in the Minnesota Star Tribue. Skip all the sentiment and hand-wringing and just jump to the statistics. Also, this is eye-opening: “By the year 2000, no large U.S. city anywhere other than on the intensely multiracial Pacific Coast had a higher share of multiracial children than Minneapolis.” Of course, the Upper Midwest and the Pacific Northwest are whiter than average regions of the country, reinforcing Steve Sailer’s point that overly large minority communities retard assimilation.
Month: July 2006
Introgression in wolves & dogs
Apropos of our discussion of evolution and dogs, and introgression, here is a new paper I stumbled upon in Molecular Ecology, Detecting introgressive hybridization between free-ranging domestic dogs and wild wolves (Canis lupus) by admixture linkage disequilibrium analysis. Linkage disequilibrium is basically the non-random association of alleles across loci. For example, imagine that you have alleles A1 and A2 at locus A, and B1 and B2 at locus B. Imagine that these two locii are on separate chromosomes (just to make it clearer, though they don’t have to be). In a randomly mating population A1 and B1 or B2 should not be correlated together in the same organism’s genome anymore than you would expected based on their frequencies. But sometimes gene-gene interactions result in the coadapted fitness of two alleles, so that A1 + B1 is more fit than A1 + B2 (imagine, if you will, higher spontaneous abortion of fetuses with A1 + B2). Nevertheless, today linkage disequilibrium is more often used to study the impact of selective and demographic forces operant upon the genome across the span of history than coadapted complexes. A strong powerful selective event on one locus, say Z, might drag adjacent regions of the genome along via a “hitch-hiking” event during the selection sweep (this is why researchers tend to look around the region coding for lactase to confirm that their technique works). As time passes recombination should break apart linkage disequilibrium, so the extent of the current associations can be highly informative. And so it is with this paper.
All the Science Blogs fit to blog (about)
On being your mother’s son
John Hawks and Tara Smith both have posts on a New York Times article describing the role of events during development on health later in life. A recent article pushed for the development of a framework to describe these kind of effects.
The basic concept is simple: during development (development in utero), the fetus recieves signals that allow it to “predict”, as it were, the environment where it will live. For example, there may be some way to sense whether the environment will be nutrient-poor or nutrient-rich. Based on this information, development proceeds in a way to favor adaptation to the predicted environment. However, if the prediction is wrong, for whatever reason, disease may result later in life. Importantly, the mother acts as the conduit for the environmental information, and may alter it to suit her needs.
The main example given in the article concerns metabolic disease like type II diabetes and obesity. The argument goes like this: due to basic physics, a fetus can’t exceed a certain size (or couldn’t, before the C-section). Thus, in a nutrient-rich environment like we have today, where a fetus would “want” to get much bigger, the mother limits the environmental signal )so as not to have a giant child), leading to a fetus that “predicts” an environment much poorer than what really exists. Once developed, the grown individual is then predisposed to gain weight. The authors put is thusly:
We argue that it is this differential rate of change between the limitations imposed by maternal constraint (which set the fetal prediction) and the reality of the enriched modern postnatal environment that has created the current high incidence of cardiovascular and metabolic disease in humans.
This is a broader framework for what some have called the “thrifty phenotype” hypothesis for the prevelance of type II diabetes, an alternative to the “thrifty genotype” hypothesis.
Baruch Spinoza, the first of “us”
Alex points me to this Rebecca Goldstein op-ed in The New York Times marking the excommunication of Baruch Spinoza. I am actually reading Goldstein’s biography of Spinoza, Betraying Spinoza: The Renegade Jew Who Gave Us Modernity, and just finished Matthew Stewart’s The Courtier and the Heretic: Leibniz, Spinoza, and the Fate of God in the Modern World. Most of you probably know the name Spinoza from Einstein’s assertion that he “believe in Spinoza’s God,” the pantheistic entity which suffused existence itself.
Update: James H. has more at The Island of Doubt.
Gould's citation record
A comment that crossed my screen via that series of tubes:
There is a perception, alluded to in the discussion above, that while Gould was a remarkably adept and influential writer of popular science, his work as a scientist per se was less notable and much less influential/respected. This is not a view universally held, as a historian of science his contributions are widely described as being outstanding, and he is said to have been very well respected in his own field of paleao biology. However I have tried to establish how influential his science was by establishing how often his scientific papers (not his popular works) have been cited in the scientific literature, through the ISI databases. For comparison, Richard Dawkins’ most highly cited scientific paper has 100 citations, Ernst Mayr’s has 173, CG Williams’ has 253 and D Tutyama’s has 394. Gould’s most highly cited paper (in Proc R Soc 1979) has 1,613 citations, and the next eight have 863, 609, 291, 169, 138, 121, 121, and 109 citations – the last of these published in 1974 is on antler size. I do not think that any claim that Gould was not highly influential as a scientist is objectively sustainable. His citation record is exceptional by any standards.
Informative and thoughtful comments welcome.
Dick Lamm speaks
I don’t blog much politics anymore because I think most of it is trivial epiphenomena. That being said, I want to point to Dick Lamm’s recent comments. Lamm says that this country is “overdue for a candid dialogue on race and ethnicity.” No shit sherlock! Back in 1996 when Lamm tried to get nominated on the Reform Party ticket I was pretty supportive because he seemed like he wasn’t much of a bullshitter. I didn’t agree with everything that Lamm promoted (Lamm changed his mind on free trade to become a skeptic, I still support free trade in the generality as a good for this nation), but his candid manner and “straight talk” (before that term was trademarked by John McCain) appealed to me. Lamm is not your typical “straight talker” on race. He is an old style environmentalist, and as a Unitarian Universalist he isn’t a rock-ribbed right-winger in the style of fellow Coloradoan Tom Tancredo (I’d be willing to bet money he’s not a theist but a humanist). Only Nixon could go to China, and only sincere progressives in the old style can transcend the excessive sentiment which seems to the consensus as exemplified by the Democratic party and the George W. Bush wing of the Repulican party when it comes to quality of life and civilizational issues. I don’t really read “political” books anymore, but I’ve just ordered Two Wands, One Nation. You’d figure with a name like Dick Lamm someone would think up a better title, but sometimes I guess we’ve got to go with substance.
Knocking out stimulant reward
I just posted this on my own blog last night and thought I’d cross-post it here, as there have been a few posts lately about neurotransmitters, neurons, and behavior.
About a month ago I saw this article about the role of the dopamine transporter in cocaine reward. For those that don’t know, the modified amino acids dopamine, norepinephrine, and serotonin, collectively known as monoamines, are neurotransmitters that are released by specific neurons in the brain and activate receptors on other neurons, sending a message from one cell to another. There are “pumps” in the membranes of the neurons that release these transmitters, which “clean up” the released monoamines so that they don’t keep activating receptors for too long. These pumps are blocked by many psychotherapeutic and recreational drugs, producing a change in brain function. While each neurotransmitter has multiple effects in the brain, the transmitter dopamine in particular is believed to participate in the behavior-reinforcing properties of both natural (food, sex, etc.) and pharmacological (drug) stimuli. Among many scientists dopamine is still believed to be a kind of “pleasure chemical” whose concentration determines the degree of positive subjective sensation produced by the environment, regardless of the specific nature of the stimulus. This idea has been called into question especially lately, though, for a number of reasons, many of which have nothing to do with this article. For instance, the effect of drugs that directly activate dopamine receptors is not euphoric in humans.
The finding that concerns us here is one made by Sora et. al. in 1998. To understand the significance of this study, it is important to know that the stimulant cocaine blocks the transporters (“pumps”) for all three monoamines. Given the assumed responsibility of dopamine for reinforcement, it has long been assumed that the block of the dopamine transporter (DAT) produces the euphoric effect of cocaine by allowing dopamine to sit around and activate its receptors longer. To test this, Sora et. al. deleted (“knocked out”) the gene encoding DAT from mice, and showed that they still prefer to spend time in a chamber in which they have previously received cocaine. This so-called conditioned place preference suggests that cocaine can act as a reward even when it cannot block DAT (because DAT doesn’t exist in these mice). Knocking out the serotonin transporter (SERT) also left cocaine reward intact (This SERT is the same as the 5-HTT mentioned in the Caspi and Moffitt study-geneticists seem to like the name 5-HTT and biochemists SERT, and some use the alternative SLC6A4 occasionally). A follow-up study showed that knocking out both DAT and SERT makes mice that do not prefer an environment they associate with cocaine. Sora et. al. took this to mean that blocking SERT is rewarding as well, which flies in the face of the fact that blocking SERT with drugs like fluoxetine (Prozac) does not produce signs of euphoria. An obvious caveat here is that the brains of DAT knockout mice are flooded with dopamine and the animals are very hyper even when they aren’t on any drugs, so findings may not generalize to normal mice.
The new study by Chen et. al. took a different approach. They found that by mutating part of DAT, they could prevent cocaine from binding to it without breaking the pump. When this mutant DAT was added back into DAT knockout mice, cocaine no longer made the mice hyperactive like normal or DAT knockout mice (paradoxically, it even calmed them) and was not rewarding. This confirms what I–and probably many other researchers–suspected was going on: the mice with DAT knocked out only showed a response to cocaine because it slightly amplified the effect of the high baseline dopamine. Possible explanations are that increased activation of serotonin receptors overcomes some negative feedback mechanism limiting dopamine levels, or that lack of DAT induces a form of plasticity in the reward pathway such that SERT blockade becomes rewarding. This still doesn’t explain other results questioning the idea of dopamine as a “pleasure chemical”, but at least it shows that cocaine, and probably methylphenidate (Ritalin) and amphetamines, do produce their reinforcing effects through inhibition of dopamine reuptake.
*I just corrected the links. For some reason the first time I posted the URLs got all messed up, even though it worked perfectly fine for my own blog when I cut and pasted from the same file on my computer.
Genotype, Recidivism and Indefinite Commitment Laws
This month Nature Reviews:Neuroscience published an opinion piece “Gene-environment interactions in psychiatry: joining forces with neuroscience” by Avshalom Caspi and Terrie E. Moffitt who follow-up on their well-cited 2002 article in Science “Role of genotype in the cycle of violence in maltreated children.” In the opinion piece the authors present a broad overview of the opportunities and challenges present in studies that address the gene-environment interactions that exist within nature. In their earlier earlier paper they:
. . studied a large sample of male children from birth to adulthood to determine why some children who are maltreated grow up to develop antisocial behavior, whereas others do not. A functional polymorphism in the gene encoding the neurotransmitter-metabolizing enzyme monoamine oxidase A (MAOA) was found to moderate the effect of maltreatment. Maltreated children with a genotype conferring high levels of MAOA expression were less likely to develop antisocial problems. These findings may partly explain why not all victims of maltreatment grow up to victimize others, and they provide epidemiological evidence that genotypes can moderate children’s sensitivity to environmental insults.
It’s quite plausible that as this science develops it will enter into the social and legal arena where it will be useful in questions dealing with Indefinite Commitment Laws. David Rose tackles this subject, and provides a useful historical overview on “genetic determinism” scaremongering, in the latest issue of Prospect (UK). I want to add to the topic, but first a little background.
Indefinite Commitment Laws are designed to keep dangerous individuals from being released from prison after they’ve completed their terms of incarceration, have been passed in 16 U.S. States and have passed through Constitutional challenges by claiming that the commitment is a medical treatment, rather than a double-jeopardy penalty of continuing incarceration. The case law can be sampled here, here and here.
The impetus for these types of laws clearly centers on fears of high recidivism probabilities of the ex-convict. Because the legal proceedings are dealing with civil commitment rather than criminal commitment, the standard of evidence must only meet a clear and convincing threshold, rather than the higher bar of beyond a reasonable doubt. Due in large part to this lower evidenciary standard the adjudicating authority becomes a de facto actuary and much of the focus deals with probability of recidivism. To gauge the boundaries of the probability estimate there are prescribed protocols which must be followed by the State. More details in this Handbook for “Sexually Violent Predator Assessment Screening Instrument for Felons: Background and Instruction” and in this Memorandum on “Civil Commitment of Sexually Violent Predators” that was prepared for Virginia Circuit Court Judges. But what are the actual probabilities of recidivism that have spurred on this legislation? In their meta-analysis of recidivism studies, Predicting Relapse: A meta-Analysis of Sexual Offender Recidivism Studies, R. Karl Hanson and Monique T. Bussiere find:
On average, the sex offense recidivism rate was 13.4% (n = 23,393; 18.9% for 1,839 rapists and 12.7% for 9,603 child molesters). The average follow-up period was 4 to 5 years. The recidivism rate for nonsexual violence was 12.2% (n = 7,155), but there was a substantial difference in the nonsexual violent recidivism rates for the child molesters (9.9%; n = 1,774) and the rapists (22.1%; n = 782). When recidivism was defined as any reoffense, the rates were predictably higher: 36.3% overall (n = 19,347), 36.9% for the child molesters (n = 3,363), and 46.2% for rapists (n = 4,017).
Margaret A. Alexander performed a meta-analysis of 79 studies that looked at recidivism rates after the criminals participated in various treatments regimes in her study, Sexual Offender Treatment Efficacy Revisited and found the following recidivism rates: Rapists (treated 20.1%, untreated 23.7%); Child Molestors (treated 14.4%, untreated 25.8%); Exhibitionists (treated 19.7%, untreated 57.1%) and the by far largest group, Types not specified (treated 13.1%, untreated 12.0%). The last entry isn’t a typo, the untreated did in fact have lower recidivism rates.
I thought it would be useful to survey the probability universe that we’re dealing with when we deprive people of their freedom via Indefinite Commitment Laws for they do work on the basis of probabilities rather than certainties and it’s quite likely that genotypic information will significantly improve the accuracy of assessments, especially when such information is combined with data on the life history of the subject. Rose summarizes the 2002 Caspi and Moffitt paper:
The paper’s hypothesis was that one of the factors that differentiates individuals’ propensity for antisocial behaviour is a particular gene-the one responsible for generating the enzyme monoamine oxidase A (MAOA). This enzyme regulates neurotransmitter levels in the brain: one of its roles is to get rid of excess serotonin, dopamine and so on, in order to keep neurological circuits working smoothly.
In fact, there are five known variants-known as alleles or genotypes-of the MAOA gene, although three of them are rare. The authors of the 2002 paper examined the two main types. The low-activity allele, which programmes the body to produce low levels of the MAOA enzyme, is found in about one third of males. The more normal, high-activity allele is found in almost all of the rest. In order to test their hypothesis about the role of MAOA, the researchers went back to the Dunedin cohort. Its members’ history had already been examined and described, so that it was already known that between the ages of eight and 11, 8 per cent of the cohort’s children had suffered “severe” maltreatment, and 28 per cent had experienced “probable” maltreatment. As we have seen, the team already knew which members of the study had exhibited antisocial behaviour, and when. Now researchers also found which of the MAOA genotypes they had by examining their DNA.
As might have been expected, the Dunedin study found that maltreatment in childhood would, on its own, make someone more likely to commit crime and display antisocial behaviour. About 35 per cent of the maltreated men with the normal high-activity genotype had shown conduct disorder, and 20 per cent had a conviction for violence. But when the two risk factors were found together-the low-activity genotype and childhood maltreatment-the correlation with antisocial behaviour was far stronger. More than 80 per cent of the men in this category had exhibited conduct disorder, and more than 30 per cent had convictions for violence. As a group, they were all among the most violent third of men. No fewer than 85 per cent
of the cohort’s men with the low-activity genotype who had also been severely maltreated went on to develop antisocial behaviour.
Notice that the rate for conviction of violent crime was 50 percent greater in the low-activity genotype group than in the high-activity genotype group. In absolute terns the former group has a conviction rate that is 10 percent greater than the latter group. Compare these rates to the the recidivism rates for rapists (18.9%), child molestors (12.7%) and violent offenders (12.2%.) When we’re dealing with probabilities of recidivism it seems that genotypic information will only serve to improve the decision process underlying indefinite commitment proceedings.
An added benefit of developing this research will be the likely erosion effect on the Axiom of Discrimination as it pertains to the question of race and crime. We already have studies which have charted monoamine oxidase activity across demographic groups:
Reported here are variations for all three demographic variables such that significantly greater enzyme activity is seen in female, older, and white subjects relative to male, younger, and black subjects.
Note that the groups most prone to low enzyme activity are males, the young, and black subjects. Another study makes the implication more explicit:
Overall, low MAO activity appears to be associated with restless and uninhibited behavior patterns, and may reflect some of the mediating effects of serotonin and sex hormones (especially androgens) on criminal behavior. Lower MAO activity is more characteristic of males than females, and appears to be lower in Blacks than Whites, and lowest during the second and third decades of life.
Of course, a research design that broaches the sensitive topic of race, genetics, and criminality is certain to dissuade many scientists from getting involved, for as Stanford’s David Botstein remarks about genetic causes of violence:
“I think there’s more scientifically to that one, a greater likelihood of finding it, more than IQ. But it’s COMPLETELY unacceptable at the moment. You can’t even talk about it. Go to any university, research center, no one — NO ONE — will talk to you about this. Why? Simple. Because of the fear that there will be a racial correlation.
We need look no further than Rose’s essay for a sampling of what the future portends:
The academy can be a compartmentalised place, with surprisingly little dialogue between disciplines, and mainstream sociological criminology is only beginning to become aware of the work described here. It may not evoke a favourable response. A recent issue of the journal Criminal Justice Matters, published by the Centre for Crime and Justice Studies at King’s College London, contained a fierce attack on the work of Terrie Moffitt and others. The article accused researchers at the Institute of Psychiatry and elsewhere of “genetic fundamentalism-a belief in a mythic, not a real genetics,” and suggested that twin studies that found a genetic component in antisocial behaviour were without value. Moffitt and her colleagues have, in fact, stressed that genetic predispositions must be “switched on” by childhood maltreatment, and that the important thing was to concentrate on eliminating this and other types of adverse environment.
Asked to give an after-dinner speech to Liberal Democrat lawyers, I caught a different glimpse of the hostility that behavioural genetic research into the causes of crime can evoke. After I had presented an account of some of the work described here, the response was viscerally critical. Speakers claimed that it was “deterministic,” and would surely lead to a wanton attack on civil liberties. One distinguished legal practitioner went so far as to demand who had funded these investigations, claiming that they must have been cooked up according to some pre-ordered, authoritarian agenda.
For most of the regular readers of this blog these types of frothing-at-the-mouth attacks are old hat. Just a few weeks ago we saw Stanford’s Barres preparing the ground for classifying science he didn’t like as hate crimes:
. . . what is the difference between a faculty member calling their African-American students lazy and one pronouncing that women are innately inferior? Some have suggested that those who are angry at Larry Summers’ comments should simply fight words with more words (hence this essay). In my view, when faculty tell their students that they are innately inferior based on race, religion, gender or sexual orientation, they are crossing a line that should not be crossed – the line that divides free speech from verbal violence – and it should not be tolerated at Harvard or anywhere else.
The Leftist Creationists who believe that humans are immune to evolutionary processes will not go down without a fight. Unfortunately, these types of creationists are a monolithic bloc within Academia and they can indeed put up impediments to research or simply resurrect the tactics used during the Sociobiology Wars. However, even if they can groom an obscurantist of Gouldian proportions, they’re going to have a difficult time refuting the work of good scientists, work which been replicated many times over. In the end these new insights should aid society in furthering the cause of justice, just as DNA testing has been instrumental in setting unjustly incarcertated people free and helping to convict those who are guilty of crimes. The science and technology are neutral but they do aid in guiding the decisions made by our arbitors of justice.
Gonna make you fall in love with…Spines
Never ever let a n* ride if you think he’s gonna slide pop’em in the spine…fo’ the money. – Bone Thugs N Harmony
The Genius is dead. Genius Re-post:
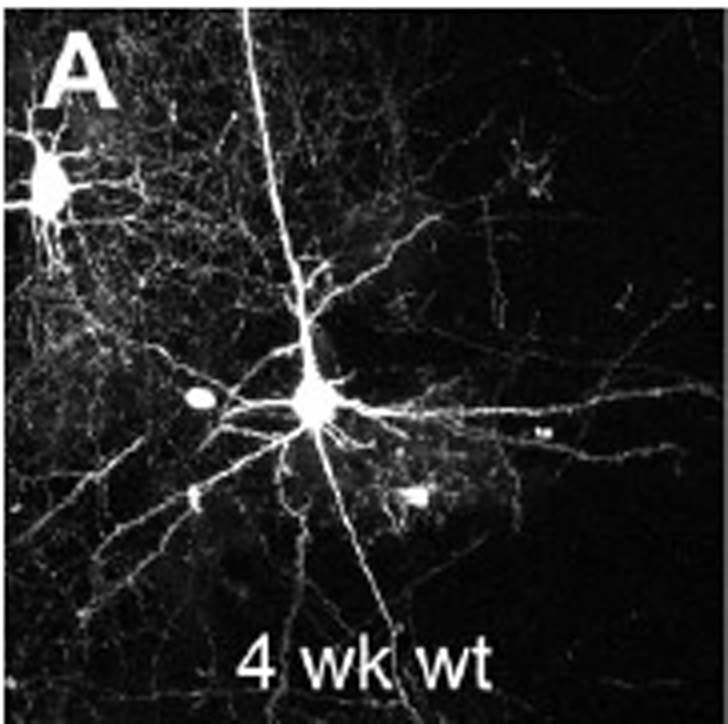
Your standard neuron in the cortex is called a pyramidal neuron. It generally has on big apical dendrite sticking out the top and a few basilar dendrites radiating around the bottom. Here’s a glowing one:
This one comes from a somewhat troublesome paper from the lab of Karel Svoboda who as far as I can tell is a guru of new imaging techniques who happens to have an interest in the details of dendrites as well. The reason why the paper was a little vexing is because the results seemed to run against a large literature showing that the major identifiable morphological problem with neurons in Fragile X Syndrome brains has to do with dendritic spines. Let me back up. Your standard neuron in the cortex has dendrites and an axon. The axon is the output structure that goes and pokes at another neuron and can fire action potentials that release neurotransmitters from its tip to send signals. The dendrites are the parts that receive signals from axons. Where an axon and a dendrite meet is called a synapse. Dendrites are studded with these little knobby-doos called spines. Spines are where most excitatory synapses in the cortex are made.
{kind=link}
Spines go through a maturation process whereby they initially come out as long wormy things called filopodia and then kind of settle back into a variety of shapes often resembling the power-up mushrooms in Super Mario Bros. or like a large kind of extended comma. The fat part at the end is referred to as the spine head and the thin part connecting to the dendritic shaft is the spine shaft.
Spine shapes change in the lifespan of the spine, but also change over the lifespan of the organism. And along side the changes in shape come changes in the number of spines per dendrite (the spine density). Since people had looked at adult human Fragile X brains and seen abnormalities with regard to spine morphology and density it seemed clear that something must be occurring during development.
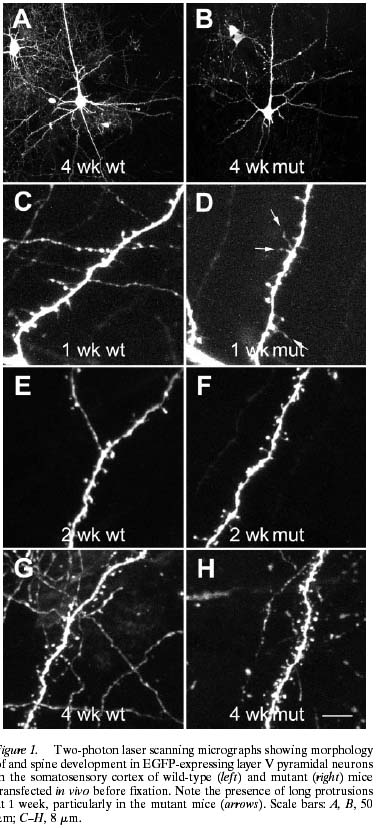
Nimchinsky et al (2001) wanted to see what happens in development so they got some normal mice and some mice lacking fragile X mental retardation protein (FMRP) that are a pretty good model for what’s happening in humans with the disorder. When the mice were one, two, or four weeks old they injected a particular part of the cortex with a fancy shmancy virus that lights up a good portion of neurons (anyone who reads science crap knows about GFP the magical jellyfish protein, that’s how they’re doing this). This allowed them to use laser microscopy to do detailed quantitative analysis on the spine lengths and density. The troublesome bit is that while they found the expected differences between FraX mice (the fragile X model) and wild-type (read: normal) mice at one week (shown below), the differences disappeared by 4 weeks. How can this be?!
They offered up a number of considerations including that they were looking at a different chunk of cortex than other people usually do and that perhaps there were some limitations to their visualization technique. The most important limitation, as it turns out, is that they can’t use their technique past about 6 weeks of age because the virus they used is too bad at infecting neurons after that. A more recent paper from the Greenough group in Illinois, who were some of the major reporters of these fragile X spine abnormalities in the first place, arrived at a resolution to this conflict.
{kind=link}
Galvez and Greenough (2005) took basically the same tack as the Svoboda group except they chose a couple of different ages. They used 25-day old mice to map onto the 4-week time point previously reported and they took another sample at age 73-76 days old. For those of you who have trouble dividing by 7, that’s about 10-11 weeks. The reason for doing this is that it is understood that a major developmental process called pruning might be at play here. Initially developing brains produce huge amounts of connections during a period of widespread synaptogenesis. They don’t need all of these connections. So they have to be ‘pruned’ back. There are lots of tree metaphors when talking about dendrites that can make it very pleasant for a neuroscientist to contemplate a tree in the park on a summer afternoon. This apparently happens in mice some time after one month of age.
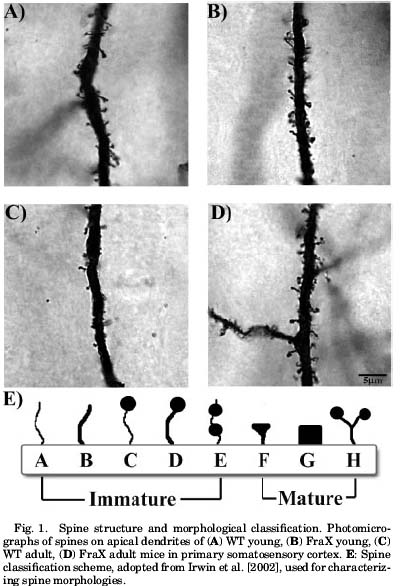
The Greenough paper used a more rudimentary staining procedure so they could look at spines in the older brains. They managed to replicate for the most part the finding that FraX mice and wild-type mice don’t differ at 4 weeks with regard to spine shape and density. But at the much later time point they differ pretty radically as illustrated here:
It appears that the major malfunction with Fragile X spines then isn’t that they can’t grow out right or anything like that. Its that they can’t be eliminated after the fact. Notice how much more bare the adult wild-type dendrite (Figure 1C from the paper) looks compared to the FraX dendrite (Fig 1D). This is awfully nice to see for two reasons. One, because it clears up an apparent discrepancy in the literature and it turns out that everyone was right which ought to make all the labs involved feel marvelous. It’s also nice because it indicates that the real neurological problems in Fragile X development start later than expected in development. I’m not quite clear on when this massive pruning event is supposed to happen in human development, but it opens a window whereby if we ever get the means to replace this protein we might ameliorate some of the effects of the syndrome.
{kind=link}
References:
Nimchinsky EA, Oberlander AM, Svoboda K (2001). Abnormal development of dendritic spines in FMR1 knock-out mice. J. Neurosci. 21:5139-5146.
Galvez R, Greenough WT (2005). Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am. J. Med. Genet. 135A:155-160.